
Fluorescence imaging of drug target proteins using chemical probes
2020-11-09 01:05HoZhuItruHmchi
Ho Zhu,Itru Hmchi,b,*
aDepartment of Synthetic Chemistry and Biological Chemistry,Graduate School of Engineering,Kyoto University,Katsura,Nishikyo-ku,Kyoto,615-8510,Japan
bERATO Innovative Molecular Technology for Neuroscience Project,Japan Science and Technology Agency(JST),Kyoto,615-8530,Japan
Keywords:
Fluorescence imaging
Drug target
Chemical probe
Ligand-directed chemistry
ABSTRACT
Fluorescence imaging can provide valuable information on the expression,distribution,and activity of drug target proteins.Chemical probes are useful small-molecule tools for fluorescence imaging with high structural flexibility and biocompatibility.In this review,we briefly introduce two classes of fluorescent probes for the visualization of drug target proteins.Enzymatically activatable probes make use of the specific enzymatic transformations that generally produce a fluorogenic response upon reacting with target enzymes.Alternatively,specific imaging can be conferred with a ligand that drives the probes to target proteins,where the labeling relies on noncovalent binding,covalent inhibition,or traceless labeling by ligand-directed chemistry.
1.Introduction
Molecular imaging allows the non-invasive and direct visualization of biological targets and assessment of biological processes in living systems.Therefore,progress in molecular imaging should contribute to the initial stages of drug development[1].It is useful to discover potent compounds to a specific protein/enzyme-ofinterest from a chemical library and it also can provide valuable information regarding biodistribution and pharmacokinetics of the drug candidates,as well as expression,localization,and activity of the drug target.Fluorescence imaging is one widely adopted imaging modality because of low cost,easy operation,and high sensitivity.The development of fluorescence imaging relies on advances in imaging probes.To date,diverse fluorescent probes have been developed,such as genetically encoded proteins,functional nanomaterials,and small-molecule probes[2-5].Chemical probes are made of synthetic small-molecule compounds that feature a high structural diversity bychemical modifications,which favors functionality and property modulation on the designer probes[5].From the standpoint of biological imaging,chemical probes label endogenous proteins without genetic manipulations,unlike fluorescent protein engineering,and they have advantages of relatively good cell permeability,low cytotoxicity,and high optical brightness with organic fluorophores when compared with fluorescent inorganic nanomaterials[2,6].
This paper is to review chemical probes for fluorescence imaging of drug target proteins,which are classified into two major groups:enzymatically activatable probes and targeted probes.Comprehensive articles have previously summarized each class of the probes[7-11].Here we mainly focus on the design principles with some representative examples,and their imaging applications are briefly introduced.
2.Enzymatically activatable probes
Enzymes are proteins that catalyze most biochemical reactions such as metabolic processes,biosynthesis,and signal transduction cascades in living systems.When a drug target is an enzyme and its substrate structure is known,activatable fluorescent probes can be rationally designed based on specific enzymatic reaction mechanisms,which allows for mapping localization of the target enzyme and assessing its activity.Enzymatically activatable probes change their emissions,generally showing a fluorescence ‘turn-on’readout signal,in response to the activities of target enzymes,which leads to increased imaging contrast and sensitivity.Several photochemical processes can be employed to achieve the optical response.
2.1.Fluorescence resonance energy transfer
Fluorescence resonance energy transfer(FRET)is one frequently used mechanism for probe design.When a fluorophore and its paired quencher were arranged in close proximity,FRET occurs to quench the fluorescence.The fluorescence can be restored by increasing the distance between them.Based on this principle,enzymatically activatable probes can be routinely designed by conjugating a fluorophore with a paired quencher through a linker of enzyme substrate(Fig.1A).Enzyme-sensitive peptides are frequently employed as the cleavable linker in protease-activatable probes,including cathepsins,caspases,matrix metalloproteinases,and furin[12-16].Such proteases are overexpressed in a number of pathologies,e.g.,cancer,allowing their use as biomarkers for tumor imaging.Small-molecule enzyme substratesarealternatively introduced as the linker.β-lactamases are a class of bacterial enzymes that efficiently hydrolyzeβ-lactam antibiotics and render antibiotics resistance in bacterial pathogens.Rao and co-workers[17,18]developed a series of fluorogenic substrates(CNIR)forβ-lactamases by linking Cy5 and QSY21 quencher through a lactam ring,i.e.,cephalosporin(Fig.1A).CNIR probes are optically dark in the native state,whileβ-lactamase cleavage triggers spontaneous fragmentation,releasing a fluorescent product containing Cy5 with a 57-fold increase in the emission intensity at 660 nm.CNIR was able to detect M.tuberculosis and quantify pulmonary infections in live mice by using β-lactamase as a reporter enzyme[18].
The FRET strategy can be readily applied to diverse fluorophores,such as near-infrared(NIR)and two-photon(TP) fluorophores which are favorable for tissue and in vivo imaging[19,20],by simply choosing their paired quenchers.However,the overall molecular size of FRET-based probes is relatively large because two fluorophores are required,which hampers their efficient cell uptake.The following strategies can be introduced for designing smaller probes.
2.2.Photoinduced electron transfer
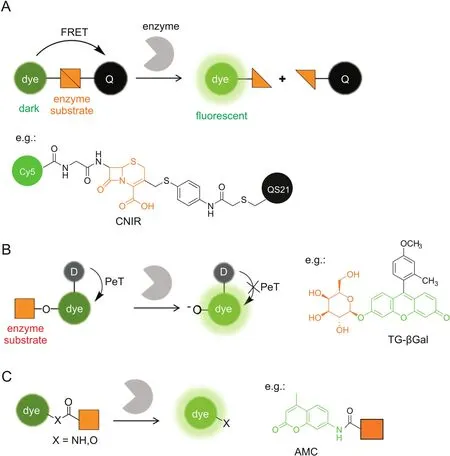
Fig.1.Enzymatically activatable probes for fluorescence ‘turn-on’imaging.The fluorescence signals are switched based on photochemical mechanisms of(A)FRET,(B)PeT,and(C)absorption alteration.For simplicity,only the ICT-based probes are illustrated in(C).Q:quencher,D:electron donor.
Photoinduced electron transfer(PeT)quenches fluorescence through electron transfer from the donor to the excited fluorophore(a-PeT)or in the opposite direction(d-PeT),which can be suppressed after binding or reaction with the target to produce a fluorescence ‘turn-on’response.PeT is frequently recruited for fluorescently sensing small biomolecules,such as the proton,metals,and reactive oxygen species[21-23],where the recognition moiety is usually electron-rich and serves as the electron donor.Urano and Kobayashi et al.[24,25]reported a novel design strategy for fluorescence probes based on the change of reduction potential of the fluorophore in a PeT system(Fig.1B).The protonated or alkylated xanthene in TokyoGreen holds a relatively high reduction potential,which favors PeT from a moderate electron donor,while the corresponding deprotonated xanthene has a decreased reduction potential,resulting in PeT inhibition and fluorescence recovery.This strategy was first exemplified by a fluorescence probe(TG-βGal)for β-galactosidase(Fig.1B)[24].TG-βGal was almost nonfluorescent.Upon hydrolysis in the presence of β-galactosidase,it became fully fluorescent.The response by TG-βGal was rapid and highly sensitive,enabling its application for fluorescence imaging ofβ-galactosidase activity in living lacZ(+)cells.In general,the PeT strategy is hardly applicable for long-wavelength,especially NIR,fluorophores.The same group exceptionally found that the fluorescence of group 14 rhodamines can be effectively regulated by PeT because of their relatively high reduction potentials[26].Very recently,a red fluorescence probe for dipeptidylpeptidase-IV has been developed based on a 2OMe-SiR600 scaffold,which produced extremely low background fluorescence for cancer detection[27].
2.3.Absorption alteration
Another strategy to develop enzymatically activatable probes relies on the alteration in their absorption spectra after encountering the target enzyme.Most of such probes are designed by modulating an internal charge transfer(ICT)process(Fig.1C).An enzyme substrate is directly attached to the electron donor of an ICT fluorophore through an amide or ester bond.After enzymatic cleavage,the free amino or hydroxyl compound is liberated with a red-shifted absorbance maximum(λmax).Enhanced fluorescence can be detected upon excitation at the red-shiftedλmax.For example,aminomethylcoumarin(AMC)is a classic fluorescent platform for protease probes,where AMC is attached at the C-terminus of a peptide substrate through an amide bond(Fig.1C)[28,29].Alternatively,when excited at the original λmaxor at the isosbestic point,one may develop a ratiometric probe that is ideally suited for the quantitative measurement.Park et al.[30]recently reported an ICT-based probe for ratiometric and TP detection of carboxylesterases in both hepatocytes and liver tissues.However,ICT fluorophores are usually sensitive to environmental polarity,which may potentially interfere with the readout of enzyme activity.
Xanthene fluorophores having a spirocyclization structure,such as fluorescein and rhodamine,are also frequently employed as scaffolds for enzymatic probes based on the absorption alteration.For example,the ‘ring-closed’form of rhodamine 110 does not absorb in the visible region and can be stabilized via amidation of the amino groups[31].Hydrolysis in the presence of the target enzyme is followed by conversion to the ‘ring-opening’form,accompanied by intense absorption and emission maxima at around 490 nm and 520 nm,respectively.Urano et al.[32]reported a rhodamine-based activatable probe(gGlu-HMRG)forγ-glutamyltranspeptidase(GGT),which involves cellular glutathione homeostasis and is overexpressed on the cell surface of several human tumors(Fig.2).gGlu-HMRG is caged with one glutamyl group and bears a hydroxymethyl group instead of the original carboxyl group.Its fluorescence kept minimal in serum containing various hydrolases over 10 min,while activation occurred within 1 min after topically spraying the peritoneal tumors in vivo.The same strategy is applicable to target ?-galactosidase based on a rhodol scaffold[33-35].Furthermore,the selenium-substituted rhodol and rhodamine are active as potent photosensitizers for enzymeactivated cell ablation[36,37].The ring-switching mechanism favors high-contrast imaging with a low background because the‘ring-closed’form principally emits no fluorescence.However,the spirocyclic structure is also sensitive to pH,and thus a proper pKavalue out of the physiological pH range is ideally required.
3.Targeted probes
Although useful,enzymatically activatable probes are limited to the imaging of particular enzymes and could readily diffuse away from the imaging target after the reaction.Targeted probes represent another class of imaging probes,which make use of specific protein recognition by a targeting ligand to accumulate probes at the protein.Compared with enzymatically activatable probes,targeted probes do not rely on enzymatic reactions and thus can cover a wider range of proteins that have a ligand-binding site.Moreover,the ligand moietyconfers increased specificity to the target protein.
3.1.Noncovalent affinity labeling
Conventional targeted probes for molecular imaging employ a straightforward strategy of noncovalent affinity labeling,which can be routinely constructed by linking a reporter tag,e.g.,fluorophore,to a targeting ligand(Fig.3A).Similar to enzymatically activatable probes,fluorogenic targeted probes that generate a fluorescence‘turn-on’response after interaction with the target protein are desirable in producing relatively high target-to-background ratios.
One strategy to generate fluorogenic targeted probes uses environment-sensitive fluorophores whose emission properties are highly sensitive to their immediate environment[38].They typically exhibit weak fluorescence in aqueous mediums but show strong and blue-shifted emissions in hydrophobic surroundings.Klymchenko and co-workers[39]reported Nile Red(NR)-derived fluorescence ‘turn-on’ligands for imaging oxytocin receptor(OTR),which is one member of GPCRs.An NR fluorophore was linked to a peptide ligand,carbetocin(CBT),through a PEG spacer which was shown to minimize the nonspecific interaction with serum proteins and lipid membranes.Ligand-receptor interaction brought the NR fluorophore into the lipid environment of the receptor,resulting in a 250-fold intensity increase and a blue-shifted emission maximum in OTR+ living cells.The fluorescence ‘turn-on’ligand was finally applied to estimate the number of OTR receptors available for ligand binding at the cell surface.
The second strategy involves intramolecular stacking between the fluorophore and the ligand,leading to a static quenching process.Ligand-protein binding induces to open the stacking complex and recovers the fluorescence.One example was reported by Peng and co-workers[40]for imaging of cyclooxygenase-2(COX-2),a common target for anti-inflammatory drugs.Indomethacin,an inhibitor of COX-2,was linked to a TP fluorophore(ANQ).A 28-fold fluorescence enhancement was detected upon binding to COX-2 in aqueous buffers.Live-cell imaging demonstrated that the COX-2 imaging probe is specifically localized in the Golgi apparatus.
Probe self-assembly was also employed for fluorescence quenching at a protein-unbound state.Hamachi’sgroup[41-43]pioneered a supramolecular approach using self-assembled nanoprobes for specific protein detection and imaging in a fluorescence ‘turn-on’manner.The nanoprobes consisting of amphiphilic ligandfluorophore conjugates can aggregate into self-assemblies in aqueous solutions,whose fluorescence is significantly quenched,whereas they can disassemble into strongly fluorescent monomers upon ligand-protein interaction.For example,HSP90,a well-known intracellular tumor biomarker,was targeted by a tetramethylrhodamine(TMR)-tethered probe carrying PU-H71 as a selectiveligand(Fig.3A)[43].TMR-PU-H71 showed a 32-fold increase in fluorescence intensity after binding with HSP90 in test tube settings.Furthermore,endogenous HSP90 expressed in live SK-BR-3 cells was fluorescently imaged by TMR-PU-H71.
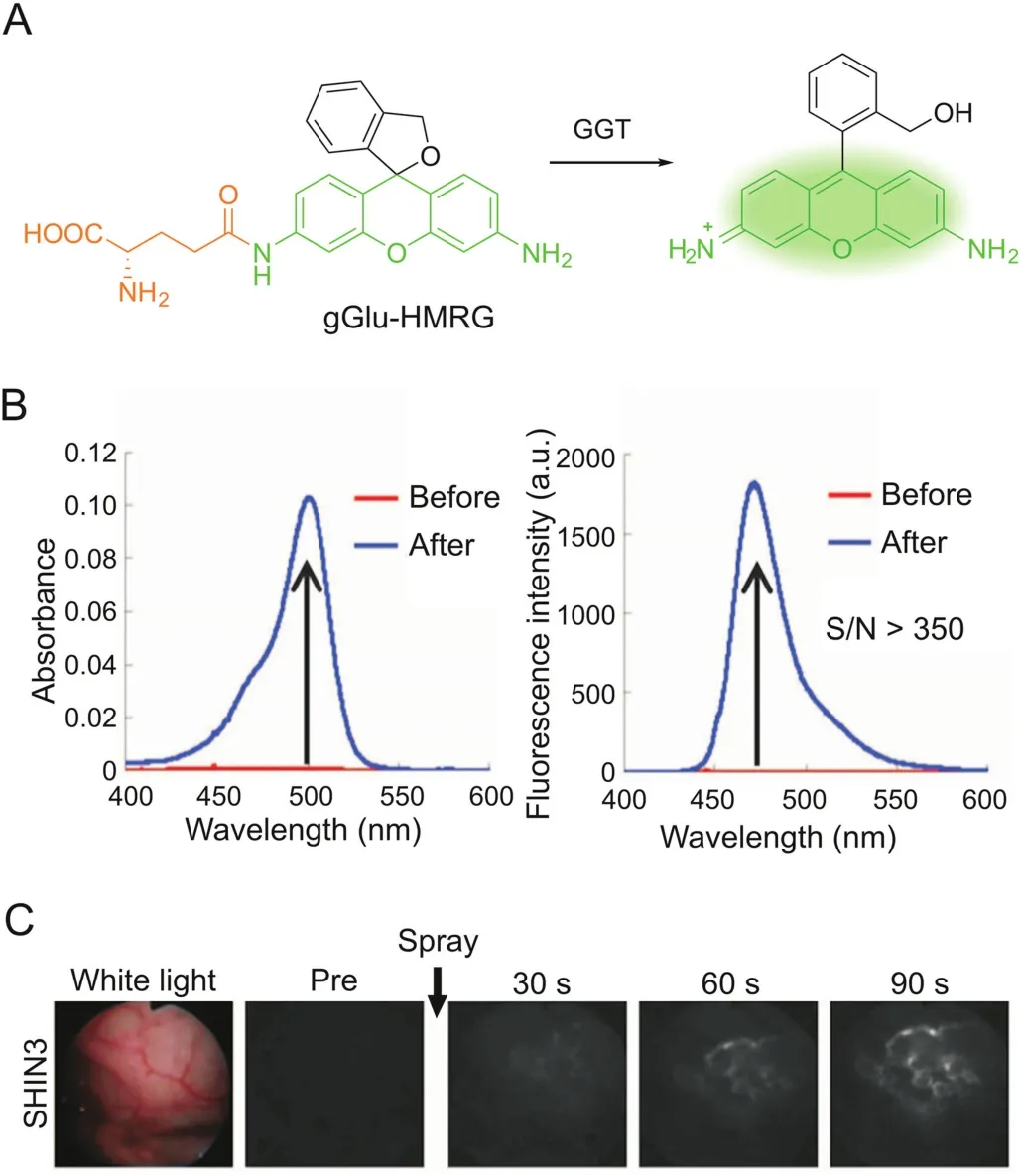
Fig.2.GGT-activatable fluorescent probe for rapid cancer detection.(A)Reaction scheme of gGlu-HMRG with GGT.(B)Changes in absorption and fluorescence of gGlu-HMRG upon reacting with GGT.(C)gGlu-HMRG illuminates SHIN3 tumors in the mouse peritoneum within 90s after spraying.Adapted from Ref.[32].Reprinted with permission from AAAS.
Noncovalent labeling probes allow rapid and reversible interactions with target proteins,which can potentially be applied for ligand-binding assays.Very recently,Hamachi and co-workers[44]have constructed a fluorescent screening system to discover positive allosteric modulators(PAMs)for GABAAreceptors(GABAARs)by using a fluorescence ‘turn-on’imaging probe.GABAAR is one major inhibitory neurotransmitter receptor in the central nervous system and is widely recognized as a clinically relevant drug target.The ‘turn-on’probe,Gzn-OG,simply consists of an Oregon Green fluorophore and a gabazine antagonist(Fig.3A),whose fluorescence is quenched by intramolecular stacking and can be recovered upon interaction with GABAARs.In live HEK293T cells expressing GABAARs,Gzn-OG emitted strong fluorescence only at the cell surface and decreased the intensity in the presence of gabazine.The ligand assay system was created by incubating Gzn-OG and GABA with GABAARs in live cells,and PAMs would increase the affinity of GABA to GABAARs,which excluded Gzn-OG from the orthosteric site and caused a decrease in the fluorescence intensity.After 4 screening steps,6 PAMs were newly identified from a library of pharmacologically active compounds(LOPAC1280).Their effects on the ion-channel activity of GABAARs were confirmed by patchclamp electrophysiological assays.
3.2.Covalent inhibition with activity-based probes
Covalent labeling after protein recognition is favorable for dynamic studies of protein localization,because it minimizes the false-positive signals by probe diffusion and ligand-protein dissociation in the cases of enzymatically activatable probes and noncovalent targeted probes,respectively.Activity-based probes(ABPs)are one representative class of covalent probes that label the active site of a defined set of enzyme targets(Fig.3B)[14,45,46].ABPs typically consist of a reactive(electrophilic)group linked to a targeting moiety and a reporter tag.Because the labeling reaction requires enzyme activity,the labeling efficiency can directly reflect the activity of given enzymes.ABPs tagged with fluorescent reporters enable imaging of the labeled targets.
Bogyo and co-workers developed a series of quenched ABPs(qABPs)as fluorescence probes for imaging of proteases(Fig.3B)[47-50].Acyloxymethyl ketone was employed as the reactive group and a quencher was attached to the acyloxy leaving group.The quenched probe becomes fluorescent upon a nucleophilic attack by a cysteine protease,which releases the quencher.They initially used a fluorophore/quencher pair of BODIPY/QSY7,which constructed a probe(GB117)that showed over 70-fold fluorescence quenching when compared with the control probe without a quencher[47].Another strength of covalent probes is the applicability to biochemical analysis for the identification of labeled targets.SDS-PAGE analysis showed highly specific labeling of cathepsin L by GB117.The qABPs strategy was adapted to target other proteases,such as cathepsin S[49]and caspase-3[50].A near-infrared fluorophore was adopted for in vivo imaging[48]and multicolor labeling of distinct cathepsins when combined with a green- fluorescent probe[49].Yao and co-workers[51]reported another class of qABPs based on the quinone methide chemistry.The covalent labeling does not inactivate the enzyme target,and thus the detection of enzyme activity produces an amplified signal,like that by enzymatically activatable probes.Fluorogenic TP imaging of enzymatic activities in live cells is demonstrated.
In addition to given enzymes,ABPs can be directed to given subcellular organelles by a moiety with specific physicochemical properties.Wright and co-workers[52]reported an acidotropic ABP(DEX-2)that selectively accumulates in lysosomes for interrogating resident enzyme activity(Fig.3B).3-(2,4-Dinitroanilino)-3′-amino-N-methyldipropylamine was incorporated for lysosome targeting and ethyl succinate epoxide was used as a cathepsinreactive warhead.Fluorescence imaging showed that DEX-2 labeled punctate vesicles in live cells,which is dependent on the acidic environment within lysosomes and the activity of target enzymes.LC-MS/MS analysis identified the enzyme targets as cathepsins B and Z and revealed increases in the enzyme activity during starvation-induced autophagy.
3.3.Ligand-directed chemistry for traceless labeling
Although powerful for specific target imaging,the labeled enzymes by ABPs lose their native activities because ABPs remain covalently bound to the enzyme active site,in the same way as irreversible inhibitors.Hamachi and co-workers[6,53]have invented new approaches to traceless protein labeling using liganddirected (LD) chemistry(Fig.3C).The target specificity is still conferred by a ligand moiety,while it is released from the protein binding pocket after labeling such that the labeled protein retains its native function.For example,ligand-directed acyl imidazole(LDAI)chemistry employs a moderately reactive alkyloxyacyl imidazole for acyl transfer from the LDAI reagent to a nucleophilic amino acid(Lys,Ser or Tyr)on the target protein surface,which liberates the imidazole fragment linked with the ligand[54].
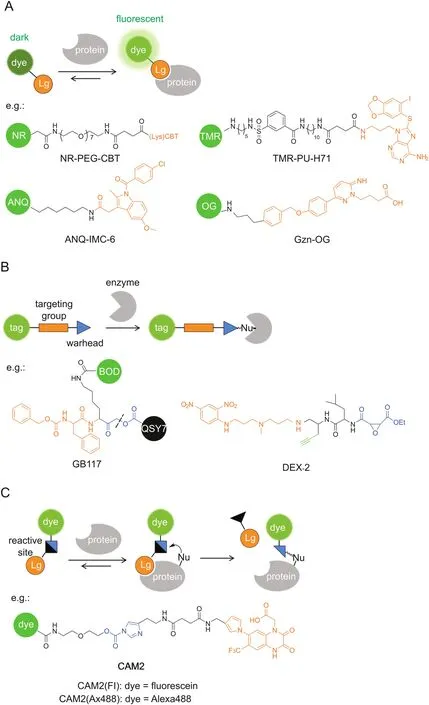
Fig.3.Targeted probes for specific imaging.The specific protein recognition is driven by a ligand moiety and the labeling relies on(A)noncovalent affinity binding,(B)covalent inhibition with ABPs,or(C)traceless labeling by LD chemistry.GB117 is one example of qABPs.The dashed line indicates the cleavage site upon cysteine attack.BOD:BODIPY,Lg:ligand,Nu:nucleophile.
The traceless labeling favors real-time monitoring of protein dynamics in the native state.Membrane proteins play fundamental roles in transport and signal transduction,and their functions are tightly regulated by protein dynamics such as protein trafficking and degradation.LDAI chemistry has demonstrated its generic applicability to various types of membrane-bound proteins on live cells,including carbonic anhydrase,bradykinin B2receptor,NMDA receptor,and folate receptor[55].Like ABPs,LD chemistry is compatible with biochemical analysis to reveal the specificity of protein labeling.The dynamics of the membrane proteins was monitored by pulse-chase analysis using LDAI chemistry,which determined the half-life of the labeled membrane proteins under almost natural cellular conditions.In addition,CLSM imaging revealed that the labeled proteins were internalized via endocytosis followed by transport to lysosomes for degradation.
LDAI chemistry has further been applied to selective chemical labeling and imaging of endogenous neurotransmitter receptors in live neurons(Fig.4)[56].Neurotransmitter receptors located on postsynaptic membranes regulate important brain functions.The AMPA-type glutamate receptors(AMPARs)are essential to synaptic plasticity and play crucial roles in memory formation.A series of‘chemical AMPAR modification’(CAM)reagents have been developed based on LDAI chemistry(Fig.3C).CAM reagents can selectively label endogenous AMPARs in both cultured neurons and brain tissues,with punctate fluorescence signals colocalized with the immunostained GluA2/3(a major subtype of AMPARs).Importantly,such chemical labeling is shown not to affect synaptic functions in brain slices by patch-clamp assays.Finally,the diffusion dynamics of endogenous AMPARs labeled with CAM reagents was analyzed by the fluorescence recovery after photobleaching(FRAP)method.
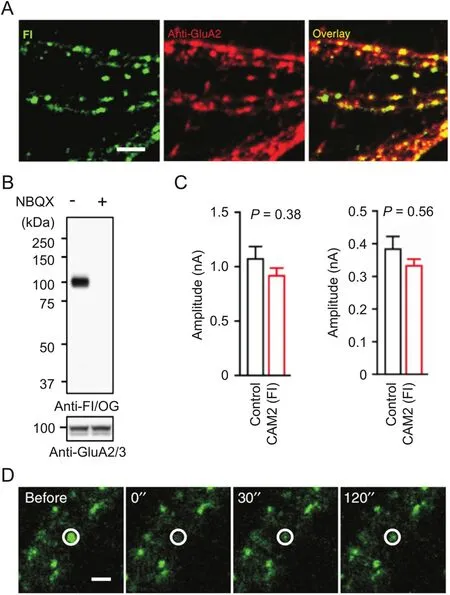
Fig.4.Chemical labeling of native AMPARs in live neurons with CAM2(Fl).(A)Colocalization imaging of CAM2(Fl)with anti-GluA2 in cultured neurons.(B)Western blot analysis of hippocampal slices labeled with CAM2(Fl).(C)Effects of chemical labeling with CAM2(Fl)on CF-EPSC(left)and PF-EPSC(right)in cerebellar slices.(D)FRAP analyses of labeled AMPARs by CAM2(Fl)in cultured neurons.
LD chemistry leaves an unoccupied ligand-binding pocket after labeling,which allows constructing a semisynthetic biosensor for the ligands.In occasional cases,the modified native protein by LD chemistry can be directly converted to a fluorescent biosensor[54,57].For example,the Ax488-tethered AMPARs prepared using CAM2(Ax488)could respond to AMPAR ligands with a fluorescence enhancement[57].Such a response is used to quantitatively analyze ligand affinity with AMPARs under live cell conditions.For a more generic design of fluorescent biosensors,the LDAI-based chemical labeling coupled with a biomolecular fluorescence quenching and recovery(BFQR)system has been developed[58].LDAI labeling first installed a fluorophore in the vicinity of the ligand-binding site,followed by fluorescence quenching by a quencher-ligand conjugate.Upon competitive binding by other ligands,the fluorescence was recovered to produce a ‘turn-on’response.The LDAI-based biosensor thus was able to discover GABAAR ligands at the benzodiazepine site by screening LOPAC1280.Two small molecules have been newly identified and performed as negative allosteric modulators for GABAARs.
4.Summary and perspectives
In this review,we have introduced two major classes of chemical probes for fluorescence imaging of drug target proteins.Enzymatically activatable probes allow for high-contrast imaging and provide a readout of the enzyme activity by virtue of their fluorescence ‘turn-on’response.They have been successfully used in the detection of bacterial infection[18]and disease biomarkers[59],organelle-specific drug delivery[60],characterization of enzyme inhibitors[61,62],etc.Targeted probes cover a wider range of proteins which are ligandable.The ligand moiety can improve the target specificity,and the following covalent labeling is favorable for dynamic imaging.Traceless labeling with LD chemistry that conserves the original protein functions should be ideally useful to evaluate the expression and dynamics of drug target proteins and investigate the drug-target interaction in a native state.Very recently,LD reagents have been conversely designed as covalent inhibitors [63],which are applicable for cell-based highthroughput ligand screening[64].
The future work on enzymatically activatable probes is likely to focus on improving the imaging resolution by enhanced enzyme specificity,increased reaction kinetics,and limited diffusion upon activation(e.g.,cell-trappable probes[37,65,66]).For targeted probes,selective visualization of intracellular proteins remains challenging.It requires good cell permeability,minimal nonspecific binding and labeling,a high-affinity ligand that can drive the probe to the target protein in a complex and crowded cytoplasmic environment.In addition,the imaging resolution could be compromised by the undesired retention of unlabeled probes inside cells,which could be addressed by design of fluorogenic probes.The future development of these chemical probes is expected to be further linked to the preclinical and clinical applications in drug discovery/development and diagnosis/therapy of various complicated diseases.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This work was funded by Japan Science and Technology Agency(JST)ERATO Grant JPMJER1802 and a Grant-in-Aid for Scientific Research on Innovative Areas“Chemistry for Multimolecular Crowding Biosystems”(17H06348).
 Journal of Pharmaceutical Analysis2020年5期
Journal of Pharmaceutical Analysis2020年5期
- Journal of Pharmaceutical Analysis的其它文章
- Catalysis-based specific detection and inhibition of tyrosinase and their application
- A pyrene-based ratiometric fluorescent probe with a large Stokes shift for selective detection of hydrogen peroxide in living cells
- Recent advances in construction of small molecule-based fluorophoredrug conjugates
- Fluorescent antibiotics for real-time tracking of pathogenic bacteria
- Strategies for PET imaging of the receptor for advanced glycation endproducts(RAGE)
- Electrochemical,spectroscopic,and molecular docking studies of the interaction between the anti-retroviral drug indinavir and dsDNA