
Recent advances in construction of small molecule-based fluorophoredrug conjugates
2020-11-09 01:05WenjieLngChonnYunLiqunZhuShuoDuLinghuiQinJingynGeShoYo
Wenjie Lng,Chonn Yun,Liqun Zhu,Shuo Du,Linghui Qin,Jingyn Ge,*,Sho Q.Yo,**
aKey Laboratory of Bioorganic Synthesis of Zhejiang Province,College of Biotechnology and Bioengineering,Zhejiang University of Technology,Hangzhou,310014,PR China
bDepartment of Chemistry,National University of Singapore,3 Science Drive 3,Singapore,117543,Singapore
cInstitute of Drug Metabolism and Pharmaceutical Analysis,College of Pharmaceutical Sciences,Zhejiang University,Hangzhou,310058,China
Keywords:
Drug delivery
Fluorescent monitoring
Prodrug
Cleavable linker
ABSTRACT
As a powerful tool to advance drug discovery,molecular imaging may provide new insights into the process of drug effect and therapy at cellular and molecular levels.When compared with other detection methods,fluorescence-based strategies are highly attractive and can be used to illuminate pathways of drugs’transport,with multi-color capacity,high specificity and good sensitivity.The conjugates of fluorescent molecules and therapeutic agents create exciting avenues for real-time monitoring of drug delivery and distribution,both in vitro and in vivo.In this short review,we discuss recent developments of small molecule-based fluorophore-drug conjugates,including non-cleavable and cleavable ones,that are capable of visualizing drug delivery.
1.Introduction
Drug discovery is a time-consuming and high-cost process,and the accurate monitoring of drug distribution and efficacy,both in vitro and in vivo,thus plays a vital role in reliable identification and optimization of drug candidates[1,2].Molecular bioimaging provides new opportunities to study drug transport in biological systems with spatial and temporal resolutions by integration with advanced techniques,such as positron emission tomography(PET)and computed tomography(CT)[3,4].Amongst the various monitoring methods,fluorescence-based approaches are quite promising owing to their non-invasiveness,real-time detection,high resolution,qualitative/quantitative capability,and can be employed in various biological systems,e.g.,animal models,tissue-based samples and single cells.Moreover,low-molecular-weight fluorescent dyes(small-molecule dyes)are particularly attractive because they are more convenient in achieving fruitful structures through chemical synthesis and can cover a wide range of wavelength and brightness[5].In recent years,small moleculebased bioimaging has been firmly established as an attractive tool for accurate monitoring of drug delivery and distribution.
In the design of fluorophore-drug conjugates,the fluorophore parts are sometimes referred to as fluorescent reporters.They can be broadly divided into non-switchable(always-on)and switchable(activatable/turn-on/ fluorogenic)versions.Compared with“always on”,the latter is more desirable as the fluorescent changes occur upon the detection,typically leading to higher signal-to-noise ratio,lower detection limit and better sensitivity.Furthermore,as shown in Fig.1,fluorophore-drug conjugates can also be categorized into two types:A)non-cleavable conjugates: fluorophores are designed to directly couple with drug molecules through covalent modifications.It is a very convenient and traditional route to track drug uptake;B)cleavable conjugates:a cleavable linker tethers the fluorophore and drug together.These linkages[6],including as ester,hydrazine,carbonate and disulfide bonds,are more labile under biological conditions or other stimuli.The latter not only enables real-time monitoring of drug delivery but also provides controllable drug release in a well-defined environment.Recently,more advances have been made to allow cleavage of the tether in order to induce change in fluorescent properties(fluorogenic/turn-on systems)or restore the drug activity(prodrug)under physiological conditions[7-9].Additionally,tumor-directing ligands such as folates,biotin,antibodies and RGD(Arg-Gly-Asp)peptides are often installed into these conjugates to form a targeted drug delivery(TDD)system[10,11].It can further reduce a drug’s cytotoxic side effects and improve selectivity in cancer treatment.These reactionbased platforms largely utilize the advantages of fluorophores and their surrounding biological environments and have recently received considerable attention from researchers.
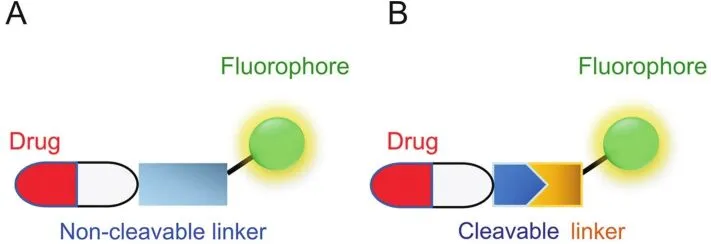
Fig.1.Schematic representation of two main types of small molecule-based fluorophore-drug conjugates.
In this short review,rather than assembling a full list of drug conjugates,we mainly focus on recent progress made in the rational constructions of these two conjugated versions,as well as their potential applications to therapeutic diagnostics including fluorogenic systems and prodrug strategies.We noted that there have already been other excellent reviews on related topics[12-16].Therefore,we mainly highlight the latest small molecular agents based on non-cleavable or cleavable fluorophore-drug conjugation strategies,not nanomaterial-based delivery systems[17-20].The prospects and future challenges of these conjugates to be used in different ways in biomedical fields are also discussed herein.It is our hope that this review will be useful for researchers seeking methods to visualize drug delivery.
2.Non-cleavable conjugates
Traditional ways to visualize drug delivery are relatively straightforward in their designs,as shown in Fig.2A,that a fluorophore is introduced at a suitable position on the bioactive molecules to form a stable and non-degradable linkage.For example,Weissleder and co-workers[21]synthesized different fluorophores,including Rhodamine Green,BODIPY-650 and SiR-COOH,to label ibrutinib(PCI-32765),and used the resulting drug conjugates to image endogenous expression of Bruton’s tyrosine kinase(Btk)from live mammalian cells.Amongst these conjugates,Ibrutinib-SiR-COOH(1,Fig.2B)is shown to possess the ideal pharmacokinetics profiles and is capable of specifically targeting Btk in singlecell imaging experiments.Recently,the Peng's group[22]reported an SLN(2,Fig.2B),containing Sulindac as both the recognition and inhibition groups and 1,9-naphthalimide as the fluorophore reporter joined by a flexible hexanediamine chain through an amineacid coupling reaction.2 is shown to be selectively distributed in three cancer cell lines(HCT-116,MCF-7 and HeLa),but not in normal cell lines(COS-7,RWPE-1 and HL-7702).With mild chemical modifications,2 is capable of inhibiting the Wnt pathway.Moreover,it acts as a good marker in xenograft tumor models to guide tumor section during surgery.
To date,fluorophores that permit excitation and emission in near infrared spectral region (~700-950 nm,I-region;~1000-1700 nm II-region)are more attractive for bioimaging in a non-invasive manner due to their excellent tissue penetration and minimal interference from biological auto fluorescence[23,24].By taking the advantage of these key attributes,Schnermann,Kobayashi and co-workers[25]used NHS ester of FNIR-774 and IR800 to conveniently synthesize conjugates of monoclonal antibodies(panitumumab and cetuximab monoclonal antibodies,respectively)in a highly efficient manner.By adjusting different dye-to-mAb ratios,the resulting conjugates showed no decrease of immunoactivity.Furthermore,compared with IR800,FNIR-774 conjugates(Fig.2C)displayed decreased liver accumulation and slightly improved tumor targeting.Burgess and co-workers[26]modified a kinase inhibitor dasatinib with a heptamethine cyanine(Cy7)NIR dye for in vivo optical imaging.The result showed that the chemical modification improved growth inhibition of cancer cells(HepG2).
However,“always-on” fluorescent conjugates lack temporal resolution.Consequently,the so-called activatable fluorescent reporters have been created to give a“switch-on”response in order to distinguish the cellular distribution of the free drugs from the conjugates(Fig.3).Efforts have been devoted to making novel conjugates based on classical sensing mechanisms.Tamoxifen(TMX)is a modulator targeting estrogen receptor(ER)and is widely used to treat breast cancer.Due to the lack of visual signals,its horizontal distributions and functions in cellular levels are not clear.Tang and co-workers[27]recently reported a novel analogue TPE-TMX(3,Fig.3B)inspired by the similarity in structures between TMX and the fluorophore tetraphenylethene(TPE).TPE is an aggregation induced emission (AIE) fluorophore,exhibiting enhanced fluorescence intensity in their aggregated states due to restricted intramolecular rotation[28,29].3,similar to TMX,can target autolysosomes in MCF-7 cells with bright signals,but not in HeLa,COS-7 and ER-negative cell lines,acting as a promising fluorescent indicator of drug distribution and cancer chemotherapy.
Hamachi and co-workers[30,31]developed a new biosensor system by using a fluorescent on/off switchable drug-based probe Gzn-OG(4,Fig.3C)to visualize γ-aminobutryic acid A receptors(GABAARs)in a real-time manner.This probe consists of Oregon Green(OG) fluorophore and a gabazine antagonist(Gzn)through a simple alkylation reaction.Upon binding of Gzn to GABAARs,the intramolecular stacking between OG and Gzn was decreased to facilitate the conformational change of 4,leading to subsequent fluorescence recovery.With the addition of GABA(agonist)to compete for binding,a significant decrease on fluorescent intensity was observed,demonstrating that 4 is a reversible fluorescent probe and would be valuable for not only imaging GABAARs but also analysis of ligands binding to GABAARs in live cells.By using this imaging-based ligand system,the authors further achieved highthroughput screening of GABAAR allosteric modulators.Their work thus successfully extended the application of fluorogenic conjugates in drug target identification and drug discovery.
Recently,Li and co-workers[32]reengineered an ibrutinib analogue IB-4(5,Fig.3D)by incorporating maleimide-coumarin moiety.Upon covalent reaction with Btk protein,fluorescence of 5 was immediately switched on due to interruption of intracellular photoinduced electron transfer(PET),allowing real-time imaging of endogenous Btk expression in live cells.Moreover,this conjugate has an alkyne handle capable of being used as an activity-based probe for subsequently target enrichment and identification[33].Moreover,our group has presented a series of imaging conjugates,based on SP600125(a non-covalent inhibitor of c-Jun N-terminal kinases(JNKs)),by using two-step labeling with biorthogonal reporter tags[34].These novel drug derivatives contain an extra built-in Michael acceptor and various bioorthogonal tags(e.g.,TCO in JP4-TCO(6,Fig.3E)).Upon incubation with cells,probe 6 first underwent covalent reaction intracellularly with endogenous JNK,which,upon further introduction of an acedan fluorophore initially quenched by tetrazine via through-bond energy transfer(TBET)mechanism[35],led to successful fluorescence turn-on and subsequent imaging of cellular JNK activities without extra washing processes.Consequently,probe 6 can be used for both in situ profiling and two-photon fluorogenic imaging of cellular enzyme activities.These successful examples,which are combined with nowashing drug imaging and chemoproteomics,have inspired researchers to develop multi-functional conjugates capable of providing more insightful information in drug discovery[36-38].
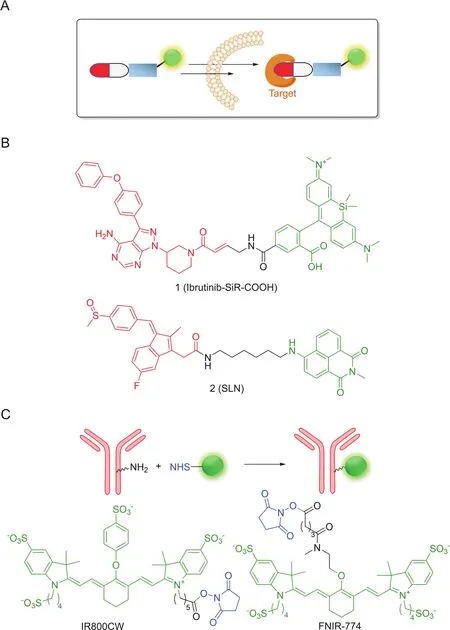
Fig.2.(A)Design principle of permanent conjugates;(B)and(C)Some representative conjugates.
Moreover,recently NIR fluorophore-based therapeutic methods such as photodynamic therapy(PDT)and photothermal therapy(PTT)have attracted considerable attention,because the fluorophores could serve not only as a visualizing module but also a drug which will be activated under light irradiation to release reactive species,such as cytotoxic reactive oxygen species(·OH,1O2,O2·-)[39,40].Cai and co-workers[41]reported a heptamethine cynanine dye IR-822 conjugated with N1-(pyridin-4-ylmethyl)ethane-1,2-diamine as a pH-sensing receptor 7(Fig.3F).Under acidic tumor microenvironment,the fluorescence of the dye switched on due to disruption of PET.Furthermore,with irradiation by a low-powered NIR laser(808 nm,0.4 W/cm2)for several minutes,the tumor temperature increased to 57°C to achieve lighttriggered PTT without extra local toxicity.
3.Cleavable conjugates
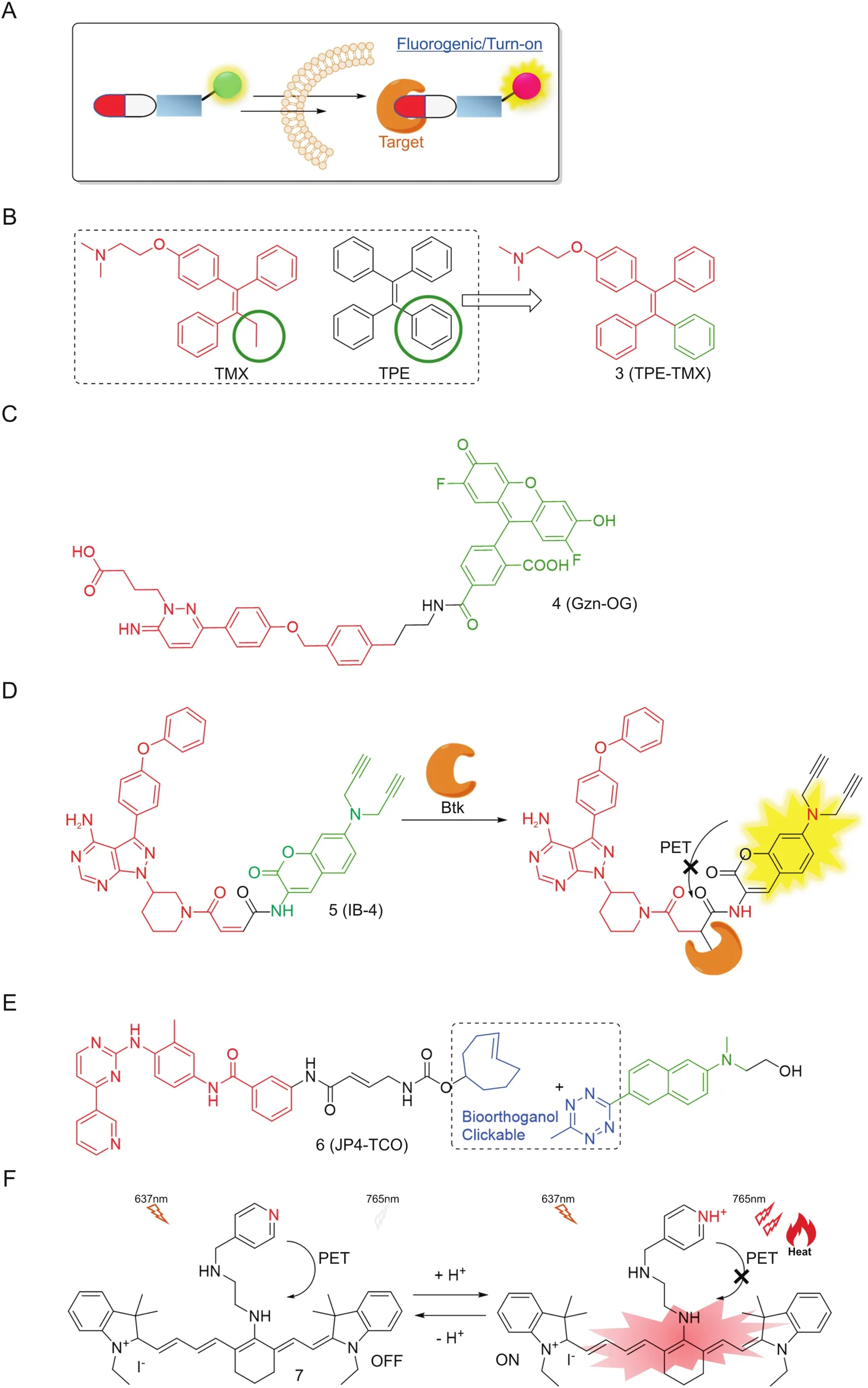
Fig.3.(A)Design principle of fluorogenic permanent conjugates;(B),(C),(D)and(E)Chemical structures of some representative conjugates;(F)NIR dye in tumor therapy.
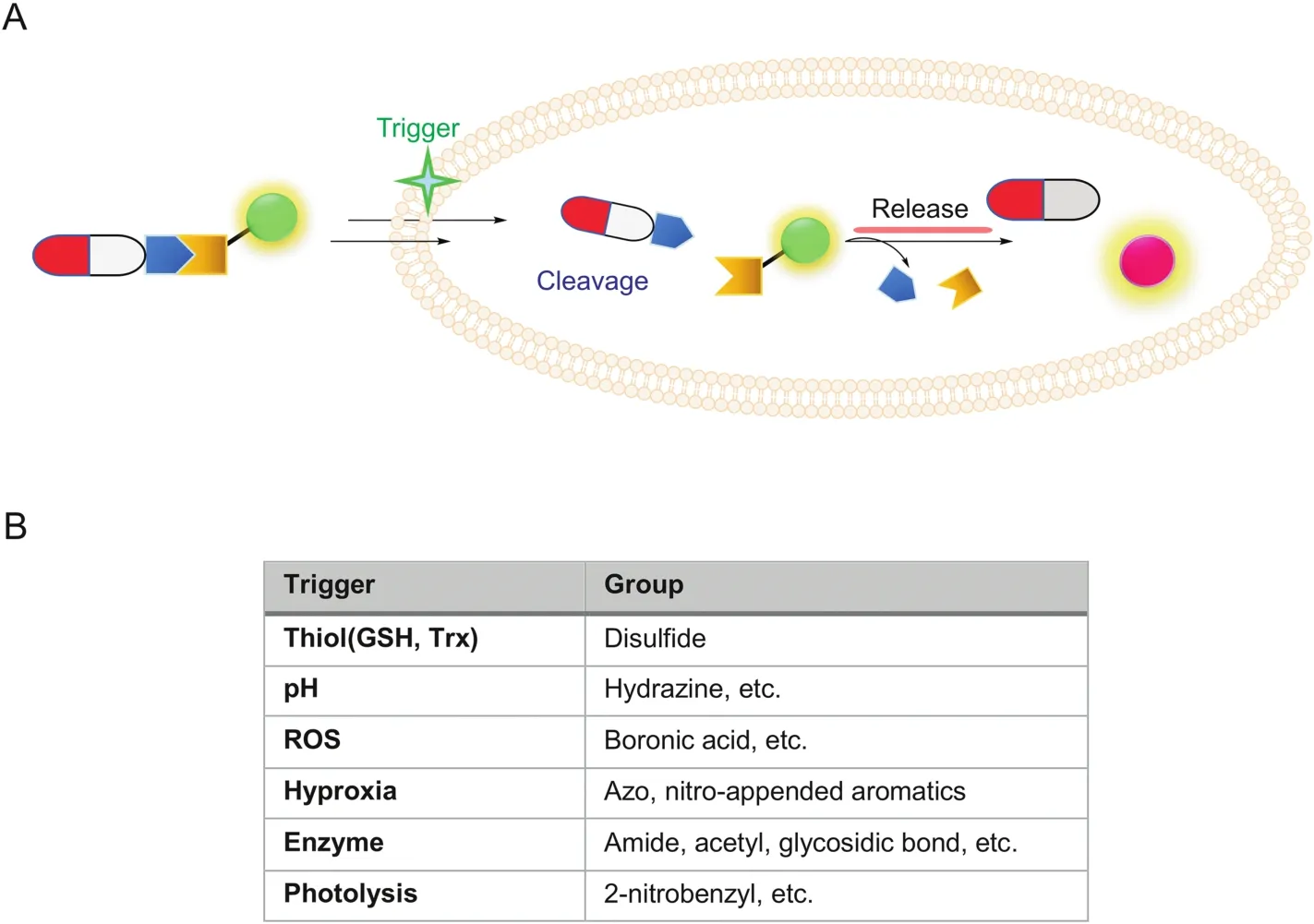
Fig.4.(A)Design principle of fluorogenic and release-based cleavable conjugates;(B)A list of some trigger conditions and cleavable linkers.
One major consideration in previous strategies is the consequence of drug modification.In some cases,the modification site can be critical to drug-target interactions.Moreover,the size of fluorophores in many cases is quite bulky and their physical properties such as solubility and hydrophobicity may affect the drugs’binding ability to the intended cellular targets.Thus,another conjugation type in which the organic dye and drug are joined together through a cleavable bond (biodegradable or external stimulus-triggered)became attractive for drug delivery.As shown in Fig.4,in most designs,upon successful cellular delivery of the cleavable conjugate,due to specific microenvironment in tumor or target cells,the conjugate could be designed to undergo controlled degradation in order to release free drug molecules(traceless release)and simultaneously generate an easy-to-monitor fluorescent signal[42,43],thus achieving “on-demand”therapy.The choice of cleavable linkers should be chosen so that they are stable during organic synthesis but highly labile(and specific)toward the expected cleavage conditions.To achieve better disease and tumor selectivity,endogenous stimuli mostly rely on abnormally expressed enzymes(esterases,matrix metalloproteinases,β-galactosidase,histone deacetylase,NAD[P]H:quinone oxidoreductase-1(NQO1))that are present in higher abundance in cancer cells[44-48],intracellular pH(acidic pH at endosomes and lysosomes),up-regulation of endogenous glutathione,hypoxia(low oxygen levels due to increased metabolic rates in tumor cells),or high levels of reactive oxygen species(ROS).External stimuli such as temperature and light have also been used[6,9].Thus,the chemical structures of cleavable linkages mainly include esters,amides/peptides,carbamates,carbonates,hydrazines,azos and disulfides.In addition,self-immolative moieties are widely used in linker design[49].In the following paragraphs,we will emphasize some linker designs,related specific cleavage mechanisms and their biological applications(Figs.5-7).
Endogenous thiol molecules such as cysteine(Cys),glutathione(GSH),hydrogen sulfide,other cysteine/thiol containing peptides,and thioredoxin(Trx)are known to be overexpressed in most cancer cells.Disulfide bonds are basically stable at extracellular environments and in the bloodstream,where only micromolarconcentrations of thiols are present.They readily undergo cleavage intracellularly through a chemical reduction process triggered by millimolar-concentration thiols in cytoplasm(e.g.,GSH)[50].Therefore,the disulfide bond plays important roles in cancer cellspecific drug design.Kim and co-workers[51]successfully developed various disulfide-based fluorescent drug conjugates,including doxorubicin,gemcitabine and camptothecin(CPT),thus allowing tumor-targeted drug delivery and bioimaging.Cleavage of the disulfide bond in conjugate 8(Fig.5A)perturbed internal charge transfer(ICT)processes and led to intramolecular cyclization to provide a naphthalimide fluorescent signal enhancement and the subsequent release of the pharmaceutically active CPT(traceless release).Besides,in this design,at the head of the fluorescent reporter,a cyclic RGD peptide(recognized and internalized by αvβ3integrin)was installed as a cancer-targeting element.Through imaging experiments and MTT toxicity assay,conjugate 8 was shown to be more active in U87 cells than in C6 cells,and was able to release the free drug within the endoplasmic reticulum(ER)of targeted cells.The non-cleavable control was shown to mainly accumulate in mitochondria.Disulfide based conjugates are widely developed to allow both release drugs and imaging[52-54].
Hypoxia is a state with abnormal low oxygen level in tissues and cells.Especially in tumor microenvironment,oxygen concentration is lowered to ~0.02%-2%(compared to ~2%-9% in normal cells).Therefore,tumor cells can also be selectively targeted by using this mechanism.Some enzymes,including flavoproteins and oxidoreductases,are specifically activated in hypoxic conditions.Azo bond,nitroaromatic heterocyclics(nitrobenzyl,nitrofuran,etc.),trimethyllocked quinone systems and indolequinone can all be dubbed as hypoxia-sensitive linkers[55-57].The Kim group[58]developed a hypoxia-response N,N-bis(2-chloroethyl)-1,4-benzene-diamine prodrug 9(Fig.5B)with an azobenzene scaffold.Rhodamine-derived azobenzene was asymmetrically synthesized and the fluorescence was quenched due to the electronic withdrawing effect of the azo bond.Besides,a triphenylphosphonium group was connected to provide mitochondrial targeting.In a hypoxic cellular environment,azoreduction led to the release of fluorescent rhodamine,allowing“switch-on”signal enhancement.This prodrug displayed good in vitro inhibition against DU145 and MDA-MB-231 under 3% O2hypoxic conditions and xenografted mouse models.Recently,Wang and co-workers[59]reported a new NIR(670/705 nm)azo-based prodrug 10(Fig.5C).It was successfully applied to the real-time visualization of drug delivery in 4T1,HepG2 cells and mice.
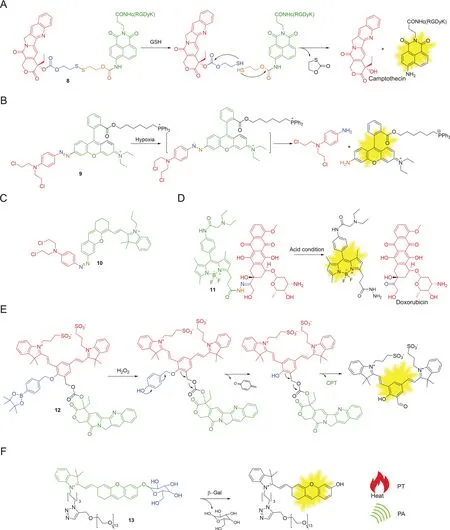
Fig.5.Some representative examples using internal cleavage conditions.
pH dysregulation is now considered to be highly related to cancer progression.Intracellular and extracellular pH in normal tissues is around 7.2,but in an extracellular tumor microenvironment is lower(6.6-7.1)[60].Hydrazone is a well-known linkage capable of creating acid-labile response.It is stable at physiological pH 7.4 but is slowly hydrolyzed in the acidic tumor environment and degrades much more quickly after entering cells due to exposure to late endosomes and lysosomes(pH 6.5-5.5)[61,62].Vendrell and co-workers[62]developed an acid-sensitive conjugate 11(Fig.5D).pH-responsive N-acylhydrazone was used to tether BODIPY and doxorubicin together.Under physiological conditions,the conjugate displayed a poor fluorescentintensity and low drugactivity.However,when 11 was located in acidic phagosomes of LPS-induced proinflammatory M1 macrophages,doxorubicin was successfully released,providing dose-dependent cytotoxicity and detectable green fluorescence.Furthermore,the conjugate could differentiate M1 microphages from others,image and modulate macrophage functions in azebrafish model.This conjugate could potentially be developed as an immunomodulatory reagent for treating immune-related diseases.
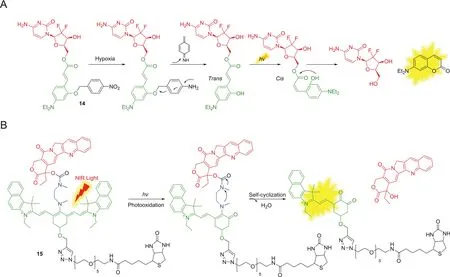
Fig.6.Some examples using photolysis.
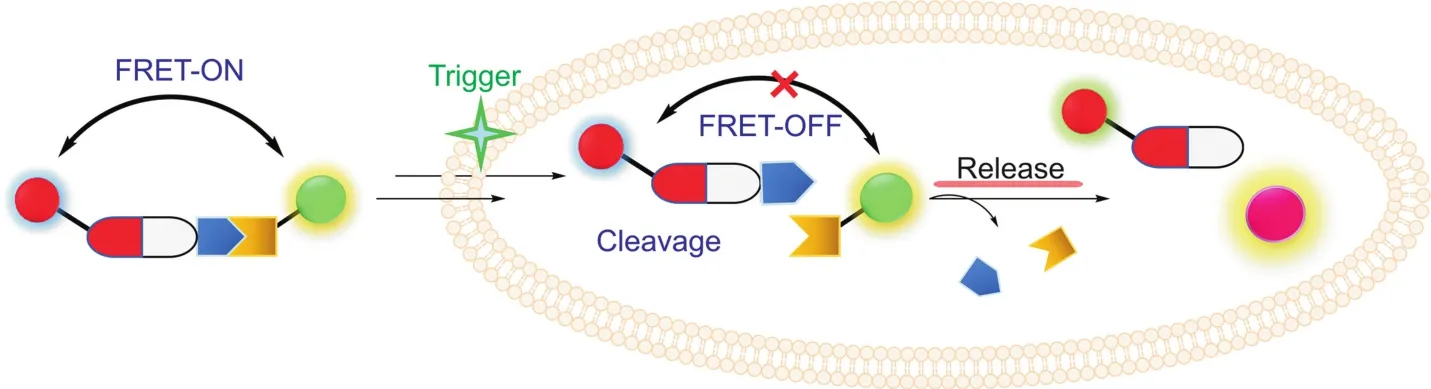
Fig.7.Design principle of FRET based conjugates.
ROSs,including hydrogen peroxide(H2O2),hypochlorous acid and hydroxyl radical play critical roles in cell signaling,cellular redox homeostasis and many other physiological processes.As ROSs are typically over-produced in tumor cells,the utilization of ROS as potential triggers for targeted drug delivery has been widely explored[63-65].For example,Shabat and coworkers[65]reported an H2O2-responsive conjugate 12(Fig.5E).The design was based on three parts:quinone cyanine-7 as the fluorophore and the central self-immolative linkage,phenylboronoic ester as a H2O2triggering substrate and CPT as the chemotherapeutic drug.Removal of the triggering substrate by H2O2generated phenolate intermediate,which subsequently underwent 1,4-elimination to release the drug molecule and a “turn-on”NIR fluorescence signal.Through intratumoral and intravenous tail vein injection in a U-87 MG tumor model,the conjugate showed a strong fluorescence signal increase in tumor region,specifically indicating the conjugate activation and drug release at the tumor site.
Many enzymes,including matrix metalloproteases and glycosidases,show different expression profiles in cancer or inflammation pathological conditions.This phenomenon has been exploited for enzyme-responsive drug release.β-galactosidase(β-gal)shows enhanced enzymatic activities in primary ovarian cancers compared with normal ovaries[66].Pu and co-workers[67]reported a β-gal-sensitive NIR conjugate 13(Fig.5F).It contains a D-galactose moiety as an enzyme trigger,a PEG linker to improve in vivo biodistribution and a NIR chromophore to provide fluorescent and photoacoustic imaging.Furthermore,after irradiation,the NIR could generate photothermal signal to further suppress cancer growth.This “many-in-one”multi-functional design has opened a new door for multimodal imaging-guided drug therapy.
Light as an external stimulus enables a biorthogonal triggered release method in which light serves as an effective activator for precise control,providing non-invasive or/and spatial-temporal drug activation in a quantitative manner[68,69].Zhang and coworkers[70]reported a sequential photo-activatable prodrug GMC-CAE-NO2(14,Fig.6A)based on hypoxia-sensitive moiety 4-nitrobenzyl group and photo-activated group.Upon nitro reduction under hypoxic conditions,the intermediate underwent 1,6-rearrangement elimination and generated a hydroxyl group at the ortho position of trans-cinnamic ester.Upon UV-irradiation,cis configuration was formed to subsequently undergo nucleophilic attack of the ester bond,leading to successfully release of the fluorescent coumarin and gemcitabine(GMC).This dual-function prodrug exhibited cytotoxicity to MCF-7 cancer cells.The limitation of this strategy is that UV-irradiation is still harmful and has poor penetration in deep tissues.Guo and co-workers[71]reported a NIR photocaged prodrug 15(Fig.6B)for precise diagnosis and therapy.A dialkylamine-based tricarboncyanine group as a light trigger was linked with CPT through a carbamate bond.The initial NIR fluorescence of 15 near 820 nm could be used to track the position of the complete prodrug.Through photo-decaging and self-elimination with temporal NIR light-irradiation,bond cleavage generated the active drug and new emission(from 820 to 535 nm),thus providing precise control on when,where and how long to offer drug treatment in vivo and ex vivo.
Moreover,single emission wavelength-based fluorophore for monitoring drug location and concentration could be affected by many factors,including photobleach of dye,probe concentration and penetration length.Ratiometric fluorescence imaging,especially based on Fo¨rster resonance energy transfer(FRET),is a powerful method and has been applied in detection of proteinprotein interactions,enzyme activities and drug deliveries[72-75].It has two different emission channels with the change of fluorescent intensity and enables us to know when and where the active drugs were delivered,and even provides quantitative information about the release of active drugs in a noninvasive manner(Fig.7).Some drugs have their own fluorescent properties,such as doxorubicin(λex= 470 nm,λem= 595 nm),camptothecin(λex= 360 nm,λem= 430 nm),SN-38(λex= 380 nm,λem= 556 nm).These drugs serve as either a FRET donor or acceptor in the design of 8,11,12,and 15.After cleavage of linkers,the pair(one is the fluorophore,the other is the drug)separate to cause the fluorescent changes of both parts.Hence,it can provide different signals from two channels to achieve better resolution.
Above all,these smart drug delivery systems aim to deliver and monitor therapeutic drugs conveniently and precisely to targeted sites at desired time,especially to cancer cells.The design concepts normally require imaging reporters,drugs and cleavable linkers which mask the fluorescence and drug activity to control“on-demand”precise drug release and imaging.
4.Conclusions
Fluorescent imaging in drug delivery,a fusion of diagnosis and therapy,is a very powerful tool providing a real-time evaluation of the efficacy of targeted drugs at a specific location.It offers direct information on drugs’distribution and accumulation(targeting ability,pharmacokinetics and dosage)to facilitate drug development through rational designs of conjugates.However,despite great advances,challenges still exist:
1)Conjugates require multiple steps of organic synthesis.The tedious purification and low yields limit scale-up processes.It is necessary to develop highly efficient and readily operable organic reactions to quickly assemble the conjugates,such as azide-alkyne clickable reactions,sulfur fluoride exchange(SuFEx)[76]and other robust reactions[77].
2)The bioavailability of fluorophores should be considered as they could bring unexpected effects to drugs’function in nonbreakable versions and also cause side effects after release in breakable versions.So during the construction of druglfuorophore conjugates,the metabolism of fluorophores needs to be analyzed comprehensively in future biological applications.
3)Besides,fluorophores not only are useful in detection but also can act as a drug to achieve good inhibitions.Yang and coworkers[78]constructed a xanthene-derivative fluorophore library and found 37 analogs with mild-to-high bactericidal activities.Some NIR dyes,including diketopyrrolopyrrole(DPP)and diiodostyryl bodipy(DBHA),belong to photosensitizers capable of drug monitoring and synergistic cancer therapy[79,80].Hence,discovery of potent multifunctional/“all-in-one”dyes,with lower tissue auto fluorescence,light scattering and photon attenuation,is noteworthy in future photo based therapy(PTT,PDT,PA,etc.).
4)Nowadays,various Janus-faced molecules in disease models have been discovered and their fluorogenic reactions are under development[81-83].It is necessary to further enlarge triggertool boxes with high sensitivity and selectivity to stimulate future drug discovery.Most triggers are specifically overexpressed in subcellular organelles.For examples,enzymes like nitroreductase,monoamine oxidases[84,85]and cytochrome C,the same as ROSs,are overexpressed in mitochondria.In addition,hydrophobic and positively charge fluorophores have preference to enter mitochondria[86,87].All these site-specific properties of fluorophore and triggers could change drugs’original targets and reduce the efficacy of drugs.As reported by Shi and co-workers[88],the modification of Ras inhibitor with heptamethine carbocyanine dye improved the original drug’s bioavailability.Hence,the integration of these modifications on“old”drugs can provide new opportunities to get infusion of fresh blood to strengthen drugs’therapeutic effects as well as providing uptake-related monitoring.
Last,although there are few successful fluorescent theranostic cases in clinical trials,it is believed that this field awaits innovations from collaborations of multidisciplinary researchers among synthetic chemists,bioengineers,biologists and others to offer better and more effective diagnostic and treatments[89-91].
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China(21708034,21877100,81903574)and Fundamental Research Funds for the Provincial Universities of Zhejiang(RF-B2019003).
 Journal of Pharmaceutical Analysis2020年5期
Journal of Pharmaceutical Analysis2020年5期
- Journal of Pharmaceutical Analysis的其它文章
- Catalysis-based specific detection and inhibition of tyrosinase and their application
- Fluorescence imaging of drug target proteins using chemical probes
- A pyrene-based ratiometric fluorescent probe with a large Stokes shift for selective detection of hydrogen peroxide in living cells
- Fluorescent antibiotics for real-time tracking of pathogenic bacteria
- Strategies for PET imaging of the receptor for advanced glycation endproducts(RAGE)
- Electrochemical,spectroscopic,and molecular docking studies of the interaction between the anti-retroviral drug indinavir and dsDNA